
Researchers Close to Understanding 'Disease Mechanisms' of ALS
Assistant Professor Carlos Castañeda leads studies of molecular causes of amyotrophic lateral sclerosis (ALS)

Researchers in the College of Arts and Sciences (A&S) are making strides in understanding the disease mechanism of amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease.
Carlos A. Castañeda, assistant professor of biology, chemistry and interdisciplinary neuroscience, and Thuy Dao, a postdoctoral researcher in chemistry, have been working with ubiquitin, a tiny molecule that tags obsolete proteins in a cell. They recently found that ubiquitin eliminates droplets of Ubiquilin-2 (UBQLN2) in solution.
The discovery is noteworthy, Castañeda says, because UBQLN2 is a protein-encoding gene, mutations to which cause ALS and various types of dementia, such as frontotemporal dementia (FTD).
“UBQLN2 is found in motor neuron inclusions of patients with ALS,” he says. “We show that UBQLN2 undergoes liquid-liquid phase separation, in which proteins coalesce into protein-rich droplets to form membraneless organelles in cells. Interestingly, dysfunction of membraneless organelle assembly and disassembly is emerging as a common pathogenic mechanism of ALS and other neurodegenerative disorders.”
The ALS Association supports the duo’s research, which in turn is the subject of a major paper in the journal Molecular Cell (Elsevier, 2018).

Other authors include Brian Martyniak G'18, a second-year Ph.D. student in chemistry and biochemistry, who belongs to Castañeda’s lab; members of J. Paul Taylor’s research group from both St. Jude Children’s Research Hospital and the Howard Hughes Medical Institute; and members of Heidi Hehnly’s lab at SUNY Upstate Medical University.
“We want to understand the mechanisms that trigger motor neurons to degenerate in ALS,” says Castañeda, the paper’s lead contact. “It appears that pathological stress granules—membraneless organelles thought to be formed by liquid-liquid phase separation of RNA-binding proteins—trigger ALS and related disorders, leading to cell death.”
Scientists know that when a eukaryotic cell is under stress, it causes certain proteins and RNA to form stress granules (SGs). While this is normal behavior, persistence of SGs or dysregulation of SG dynamics can promote disease states. Castañeda and Dao, in collaboration with Taylor’s group, showed that UBQLN2 was “recruited” to SGs. “This gives our work potential ALS relevance, since mutations in UBQLN2 might lead to defects in either SG assembly, or SG disassembly, or both,” Castañeda says.
There are billions of neurons, or nerve cells, in the nervous system. (The brain alone has more than 100 billion of them.) When ALS attacks neurons, their corresponding muscles weaken and die. People with ALS eventually lose the ability to speak, eat, move or breathe.
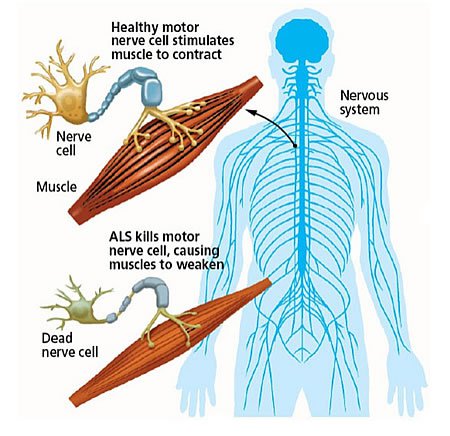
Castañeda, who studies proteins associated with neurodegenerative and neuromuscular disease, explains that muscle weakness or stiffness is usually the first sign of ALS: “It is followed by atrophy and paralysis of the muscles of the limbs and trunk, and of the muscles controlling vital functions. The average survival time is three years after diagnosis.”
Ubiquitin and UBQLN2 are part of what of Castañeda calls a “quality-control mechanism,” which maintains proteins at their proper levels during the lifespan of a cell. (Unlike other cells, which live several days or weeks, neurons typically last an entire lifetime.) Any kind of disruption to protein homeostasis usually impairs neuronal development and function.
“We postulated—and eventually confirmed with microscopy and nuclear magnetic resonance spectroscopy—that ubiquitin disrupts UBQLN2 liquid-liquid phase separation,” says Castañeda, who joined Syracuse’s faculty in 2014. “This was significant because ubiquitin tags many proteins, at one point or another.”
Castañeda ultimately hopes to redirect UBQLN2 out the “ubiquinated” substrates in SGs and into protein quality-control pathways. “UBQLN2 is like a shuttle, ferrying misfolded proteins to the cell’s protein-recycling plant,” he continues. “Under normal conditions, SGs dissipate when the stress condition is removed. However, if the condition impairs SG assembly in any way, ALS-linked RNA-binding proteins begin to aggregate.”
While there is no cure for ALS, many people with the disease live longer, thanks to clinical management and two FDA-approved drugs: riluzole and radicava.
Castañeda is optimistic his innovative work with ubiquitin and UBQLN2 will achieve a greater understanding of ALS’ molecular mechanisms and lead to a cure. “UBQLN2 interacts with other RNA-binding proteins, including TDP-43, which is found in 97 percent of inclusions of patients diagnosed with familial or sporadic ALS,” he says. “I look forward to investigating these interactions.”
Studies show that most people who develop ALS are between the ages of 40 and 70, with the disease being 20 percent more common in men. Although scientists struggle to determine the specific genetics or environmental factors that trigger ALS, they find that military veterans, particularly those deployed in the Gulf War from 1990-91, are twice as likely to develop the disease.
“Defects in protein recycling contribute to neurodegeneration,” Castañeda says. “The more we understand UBQLN2’s biological functions—specifically, how its mutations lead to ALS—the better able we can develop new therapies.”
The ALS Association is the only national nonprofit organization fighting Lou Gehrig’s Disease on every front. By leading the way in global research, providing assistance for people with ALS through a nationwide network of chapters, coordinating multidisciplinary care through certified clinical care centers and fostering government partnerships, the association builds hope and enhances quality of life while aggressively searching for new treatments and a cure.